Skip to main content
Search
Search This Blog
Trends in Science
Pages
Home
More…
Posts
Showing posts from 2010
Show all
December 24, 2010
Xmas holiday: fun videos
November 23, 2010
Reading with your tongue
November 13, 2010
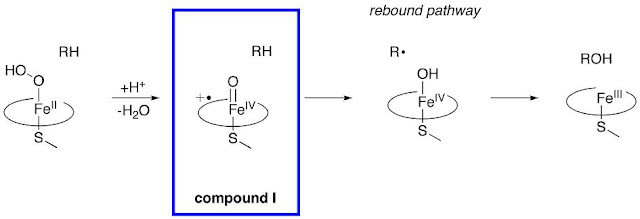
P450 Compound I observed
October 29, 2010
Hidden code in proteins
October 17, 2010
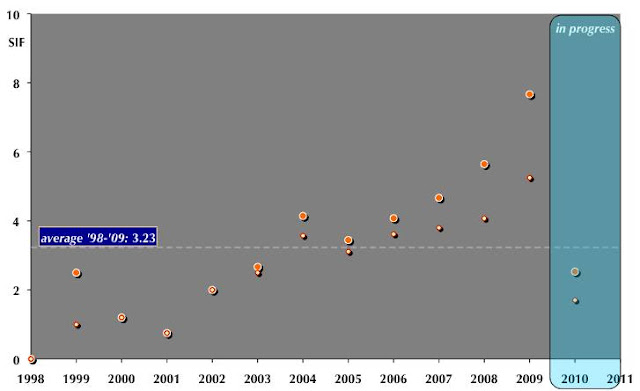
Scientist Impact Factors (SIF)
October 15, 2010
Sílvia Osuna leaving for the States
October 04, 2010
Lost correspondence of Francis Crick
October 02, 2010
SSB-D functional in NWChem 6.0
October 01, 2010
Premi Extraordinari de Doctorat for Mireia Güell
September 08, 2010
Intra- and intermolecular dispersion
August 03, 2010
Popularity poll density functionals 2010 (DFT2010)
July 22, 2010
Importance of dispersion energy for DFT methods
June 17, 2010
Spin-state-corrected GTOs
June 15, 2010
IXth Girona Seminar
May 25, 2010
IQC at Science Park
May 23, 2010
Minimum polarizability principle of spin states
May 08, 2010
Fullerenes at the Temps de Flors
May 05, 2010
Magnetizabilities with SSB-D
April 21, 2010
Good performance of SSB-D for NMR
April 16, 2010
CFOUR in parallel
April 15, 2010
Mikael Johansson starts as Juan de la Cierva
March 26, 2010
Dr. Sílvia Osuna
March 21, 2010
SSB-D functional in NWChem
March 20, 2010
Occupations of irreps in NWChem
March 12, 2010
My smart converter
February 11, 2010
CFOUR
February 11, 2010
Trends in science
Newer Posts
Home